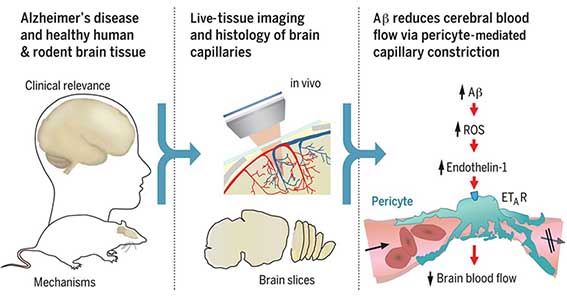
アルツハイマー病(Alzheimer's disease: AD)では、脳血流量(cerebral blood flood: CBF)が低下している。そのメカニズムはどうなっているのか。
英ロンドン大学のNortley, Attwellらは、毛細血管の径を調整するペリサイトに何等かの障害が起こっている可能性を考え、ヒトやげっ歯類の脳組織でペリサイトの動態を調べた。

Aβが脳内血管を収縮させる
まず、著者らは、脳腫瘍の評価目的で切除したヒトの生検検体から、正常組織部分を切り出し、それをスライスカルチャーした。
その脳組織をノルアドレナリン、グルタミン酸などで処置すると、ペリサイトは毛細血管を収縮/拡張させ、系がworkする事を示した。
次に、リコンビナントAβをオリゴマー化して、脳組織に処置すると、毛細血管はペリサイトの細胞体近くで径が減少し、ペリサイトが毛細血管を収縮させていると判断した(Km 4.7nM)。
Aβモノマーではこの現象は起こらなかった。
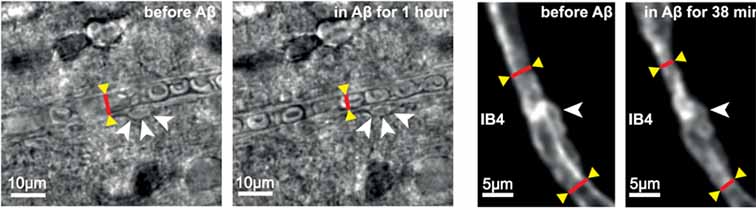
ヒト検体には限りがあるので、ラットの脳で同様の実験を行った。
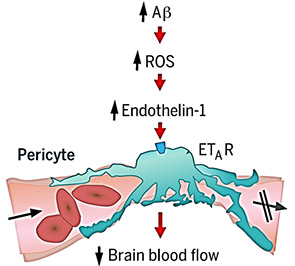
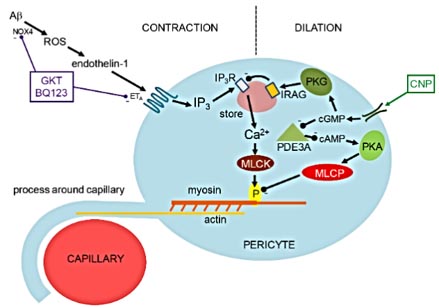
活性酸素(Reactive oxygen species: ROS)スカベンジャー(SOD1)、NADPHオキシダーゼ(NOX)阻害剤(GKT, DPI)でこの現象はブロックされ、Aβオリゴマーは、活性酸素産生を上げる事で、血管収縮に関与していると考えられた。
また、エンドセリン処置によって毛細血管は収縮し、この作用はエンドセリンA受容体(ETA)阻害剤(BQ123)でブロックされた。ROSスカベンジャー投与はこの作用をブロックせず、ETA阻害剤はH2O2による血管収縮作用をブロックしたことから、「Aβオリゴマー → ROS↑ → エンドセリン1 → ETAを介したペリサイトの血管収縮」というメカニズムが考えられた。
Dehydroethidium(DHE)でROS産生細胞を調べると、DHEはほとんどペリサイトのマーカーと一致した。
また、ヒト脳組織でAβプラークありとなしの脳を比較すると、Aβプラークありの脳では、ペリサイトの細胞体近くで血管系が減少していた。
ヒトAPPノックインマウス(APPNL-G-F)とNG2-DsRedマウスを交配し、Aβプラークとペリサイトを可視化して二光子顕微鏡で観察したところ、このマウスではヒト脳と同じようにペリサイトの細胞体近くで血管系が減少していた。
Aβプラークのない小脳では、血管系は野生型と変わらなかった。また、細動脈や細静脈の血管系は野生型と変わらなかった。
最後に、これらの減少が、薬剤投与によりブロックできるか検証した。
ラットスライスカルチャーにAβオリゴマーを処置し、NOX4阻害剤(GKT)とETA阻害薬(BQ123)の組み合わせを試したところ、血管系の減少は若干抑えられた。
ナトリウム利尿ペプチドC(C-type natriuretic peptide: CNP)を処置すると、血管系の減少はほとんど抑えられた。
これらの薬剤は、ADのCBF減少を抑える効果が期待できる。
Like a computer, the brain needs a reliable source of power, which is provided as oxygen and glucose in the blood. However, in many neurological disorders this energy supply is disrupted. Brain blood flow is controlled by adjustment of the diameters of the vessels supplying the blood. Nortley et al. found that, both in humans developing Alzheimer's disease (AD) and in a mouse model of AD, brain capillaries become squeezed by pericytes (see the Perspective by Liesz). By defining the underlying mechanism, they suggest potential targets for therapy in early AD. Science , this issue p. [eaav9518][1]; see also p. [223][2] ### INTRODUCTION In Alzheimer’s disease (AD), the production of amyloid β (Aβ) oligomers and downstream tau dysfunction are thought to cause neuronal damage, in particular a loss of synapses and synaptic plasticity, which results in cognitive impairment. However, epidemiological data show that vascular factors are important contributors to AD risk, and biomarker research has shown that the first change in AD is a decrease of cerebral blood flow. Because most of the vascular resistance within the brain is located in capillaries, this could reflect a dysfunction of contractile pericytes on capillary walls. Indeed, pericytes are known to regulate cerebral blood flow physiologically and to severely restrict blood flow after stroke. ### RATIONALE We examined the role of pericytes in Alzheimer’s disease by examining cerebral capillaries in humans and mice developing AD, and by applying Aβ to capillaries. We used freshly fixed brain biopsies from cognitively impaired living humans who were depositing Aβ plaques, and also carried out in vivo imaging in a knock-in mouse model of AD. We measured capillary diameters at positions near pericytes in order to assess whether the capillaries became constricted in AD, because this would lead to a decrease of cerebral blood flow and hence a decrease of the glucose and oxygen supply to the brain tissue. In addition, to investigate one mediator already thought to be important in AD, we applied Aβ to human brain slices made from normal tissue that was removed from patients undergoing neurosurgical glioma resection, as well as to rodent brain slices. Aβ was applied in the oligomeric form, which is thought to contribute to cognitive decline. This allowed us to examine whether Aβ itself might alter cerebral blood flow, and to use pharmacology to investigate the mechanism of any such effect. ### RESULTS Both in humans developing AD and in the mouse model of AD, capillaries were constricted specifically at pericyte locations, but arterioles and venules were unchanged in diameter. Thus, the reduction of cerebral blood flow known to occur in AD is produced by capillaries rather than by arterioles. The capillary constriction increased rapidly with the severity of Aβ deposition, and we calculated that in the human cortex this constriction would have the effect of reducing cerebral blood flow by approximately half; this is comparable to the decrease of blood flow measured experimentally in affected parts of the AD brain. In the AD mouse cerebellum, which lacks Aβ deposition at the age examined, there was no capillary constriction, supporting the idea of a causal link between Aβ level and constriction of capillaries. Aβ itself was found to constrict both human and rodent capillaries through a mechanism involving the generation of reactive oxygen species (ROS), mainly by NOX4 (reduced nicotinamide adenine dinucleotide phosphate oxidase 4). The ROS then triggered the release of endothelin-1, which acted on ETA receptors to evoke pericyte contraction, thus causing capillary constriction. The Aβ-evoked constriction could be halted by blocking NOX4 and ETA receptors, and was reversed by applying the vasodilator C-type natriuretic peptide. ### CONCLUSION These data reconcile genetic evidence for a role of Aβ in triggering neuronal damage and cognitive decline in AD with the fact that a decrease of cerebral blood flow is the first clinically detectable change in AD. They imply that attention should be given to vascular mechanisms in AD as well as to signaling pathways that act directly on neurons or glia, and suggest novel therapeutic approaches for treating early AD by targeting drugs to brain pericytes. Our findings also raise the question of what fraction of the damage to synapses and neurons in AD reflects direct actions of Aβ and downstream tau, and what fraction is a consequence of the decrease of energy supply that Aβ produces by constricting capillaries. ![Figure][3] Live human and rodent brain capillaries become constricted in Alzheimer’s disease. Tissue from humans and rodents (left) that were healthy or developing Alzheimer’s disease (AD) was imaged in vivo and as brain slices (center), revealing that pericytes constrict brain capillaries early in AD via a mechanism involving ROS generation and release of endothelin-1, which activates ETA receptors (right). Cerebral blood flow is reduced early in the onset of Alzheimer’s disease (AD). Because most of the vascular resistance within the brain is in capillaries, this could reflect dysfunction of contractile pericytes on capillary walls. We used live and rapidly fixed biopsied human tissue to establish disease relevance, and rodent experiments to define mechanism. We found that in humans with cognitive decline, amyloid β (Aβ) constricts brain capillaries at pericyte locations. This was caused by Aβ generating reactive oxygen species, which evoked the release of endothelin-1 (ET) that activated pericyte ETA receptors. Capillary, but not arteriole, constriction also occurred in vivo in a mouse model of AD. Thus, inhibiting the capillary constriction caused by Aβ could potentially reduce energy lack and neurodegeneration in AD. [1]: /lookup/doi/10.1126/science.aav9518 [2]: /lookup/doi/10.1126/science.aay2720 [3]: pending:yes

My View
アルツハイマー病(Alzheimer's disease: AD)では脳血流量(cerebral blood flood: CBF)が40%も減少しているという報告があります(Asllani et al, JCBFM, 2008)。
また、AD脳の毛細血管を病理学的に解析すると、老人斑と呼ばれるアミロイドβ(Amyloid β: Aβ)プラークの近くで毛細血管が狭小化したり、血流が途絶えたりしています(Kitaguchi et al, Neurosci Lett, 2007; Hunter et al, PLOS ONE, 2012)。
脳内の血流低下はADの認知機能低下の増悪因子になると考えられます。
このような毛細血管の変化のメカニズムはどうなっているのか、また、この現象は改善可能なのか、つまり、治療対象となり得るのか、という事をテーマに研究されています。
ここのラボでは、以前からペリサイトと呼ばれる壁細胞が、微小血管を拡張・収縮させ、血流調節を行っている事を報告してきました(Hall et al, Nature, 2014)。
今回もペリサイトが鍵だと考え、実験を進めています。
ペリサイトは、Neurovascular couplingに寄与していたり、脳梗塞や心筋梗塞で、血栓解除後に毛細血管の再灌流を妨げる”no-reflow phenomenon”の原因となっていたりと、血管系の分野では以前から注目されていました(Yemisci et al, Nat Med, 2009; Hall et al, Nature, 2014)。
さらに、ペリサイトの障害はAD病理を悪化させる(Sagare et al, Nat Commun, 2013)、早期ADや軽度認知機能障害の脳内でペリサイトの障害と血液脳関門(Blood brain barrier: BBB)の破綻が起こっている(Nation et al, Nat Med, 2019)等、ADに関係する研究も次々と報告されており、ADの治療ターゲットとしても注目されています。
以前から、Aβが酸化ストレス→エンドセリン1を介して毛細血管の収縮に寄与する事は報告されており(Deane et al, Nat Med, 2003; Deane et al, J Clin Invest, 2012)、「毛細血管収縮といえばペリサイト」という事で、それらのパーツを組み合わせて今回の結果が導き出されたように思います。
ちょっと穿った見方をすると、APPNL-G-F;NG2-DsRedマウスで、活性酸素産生細胞がほとんどペリサイト、という部分はちょっとやりすぎのように思いました。もちろんペリサイトもだとは思いますが、ほかの細胞だってAβによってROS産生は誘導されますし、免疫系細胞はどーなってるの?と突っ込みたくなります(Supplementaryで少し触れられていますが)。
あと、最後にAβによる血管収縮を防げる薬剤の候補が出てきましたが、ADモデル動物に投与して脳血流が改善したかどうかetc.は見ていないので、the jury is still out...
続報を待ちます。