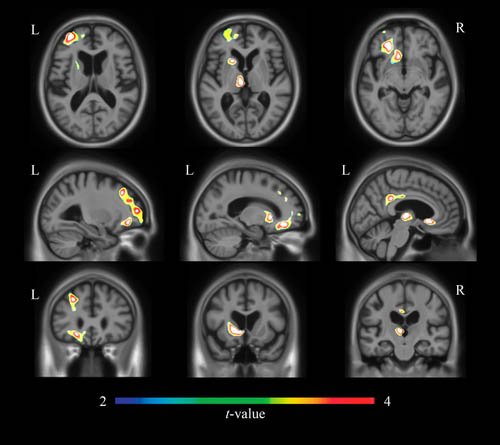
筋萎縮性側索硬化症(Amyotrophic Lateral Sclerosis: ALS)の多くの症例と、前頭側頭型認知症(Fronttemporal dementia:FTD)の約45%の症例でDNA-binding protein 43(TDP-43)の機能障害が確認されている。
また、家族性ALSの症例の中にTDP-43遺伝子の変異も確認された。
しかしながら、TDP-43の異常と神経変性を結ぶ機序は今だ不明である。
カリフォルニア大学のMelamedとDon Clevelandらは、核内のTDP-43の機能喪失によりスプライシング異常がおこり、それによるstathmin 2のmRNAの喪失が神経障害につながる事を明らかにした。

TDP-43による神経変性にstathmin-2が関与する
TDP-43の欠失/変異はstathmin-2の発現を抑制する
彼らは、まずヒトの神経細胞を用いて、
- siRNAでTDP-43を欠失
- CRISPER/Cas9で内因性のTDP-43にALS変異を挿入
を行った。
1⇒ RNAの解析を行ったところ、TDP-43は1/4程度まで減少し、この操作で最も影響を受けたmRNAは、神経の成長に関与するstathmin-2だった。
2⇒ RNA seqで野生型と変異型を比較したところ、950種類のmRNAに変化があった。stathmin-2 mRNAは、1.7倍減少しており、タンパクレベルではTDP-43N352S変異で2倍の減少だった。
ALS患者由来の神経細胞ではstathmin-2が減少している
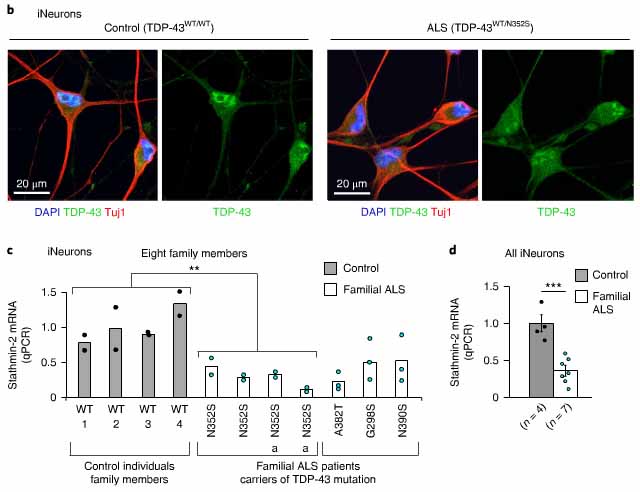
ALS患者(4人はTDP-43N352S変異、4人は変異無し)の線維芽細胞から神経細胞に直接分化させたところ、stathmin-2のmRNAレベルは50%程度減少していた。
線維芽細胞の段階では、TDP-43は核内に限局していたが、神経細胞に分化させると、TDP-43は細胞外に蓄積し、TDP-43N352S変異ではその現象がより顕著だった。
他の家族性変異のある患者(TDP-43G298S、TDP-43A382T、TDP-43N390S)から採取した線維芽細胞由来の神経細胞でも、同様にstathmin-2のmRNAが減少していた。
TDP-43はstathmin-2のpre-mRNAのポリアデニル化を抑制する
stathmin-2遺伝子には5つのエクソンがあり、エクソン4とエクソン5の間に選択的エクソン(選択的スプライシングを受けるエクソン)がある。
しかしながら、TDP-43を減少/変異させると、新たな選択的エクソンが誘導され、RNA-seqで解析するとそれはイントロン1の部位に出現していた。この新たなエクソン(エクソン2a)は、野生型の細胞では存在せず、TDP-43の欠失か、TDP-43N352Sの変異挿入で出現した。またその際、stathmin-2タンパクの減少を伴っていた。
このエクソン2aの組み込みが選択的ポリアデニル化を促す可能性を考え、anchored oligo (dT)プライマーを用いてRT-PCRを行ったところ、エクソン2aを末端としてポリアデニル化がおこっている事を確認した。
エクソン2aにはポリアデニル化シグナル(AUUAAA)があった。SH-SY5Y細胞に変異TDP-43を導入すると、エクソン2aを含むstathmin-2のpre-mRNAが出現し、全長のmRNAは減少していた。
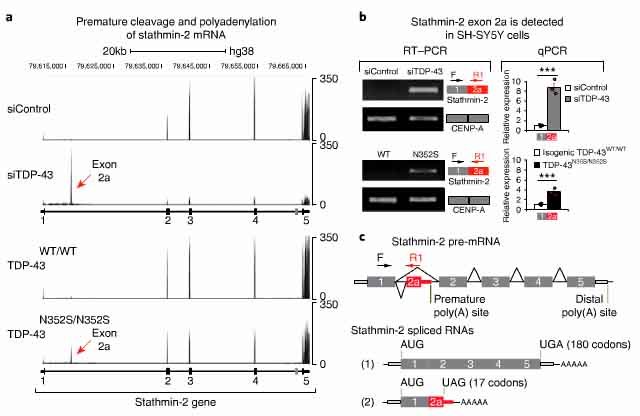
エクソン2aには、3' スプライシングサイトの下流に三カ所のTDP-43結合サイト(UGUGUG)があり、iCLIPで実際にTDP-43が結合する事を確認した。
これらの結果から、TDP-43が減少すると、通常はイントロン1にあるポリアデニル化サイトが使われ、stathmin-2のmRNA前駆体が切断され、機能的なstathmin-2の発現が抑制される事が考えられた。
stathmin-2のmRNAは主に運動神経に発現している
過去文献でstathmin-2の局在を調べると、stathmin-2は主に運動神経に発現しているようであった。腰髄の運動神経細胞をレーザーマイクロダイセクションで切り取りRNAシークエンスを行ったところ、stathmin-2のmRNA量が20倍近くあり、4種類のstathmin遺伝子の中でもstathmin-2が最も神経細胞内で多くなっていた。
切断されたstathmin-2と、全長stathmin-2の減少はALS患者の運動神経内で確認される
レーザーマイクロダイセクションで切り取った腰髄運動神経細胞のstathmin-2遺伝子を調べたところ、全てのALS患者(n = 13)ではエクソン2aを確認し、健常人(n = 7)では確認されなかった。
また、C9orf72のGGGGCCリピート配列の延長を持つALS患者(n = 3)でこの現象を認め、健常人(n = 8)や、SOD1変異患者(n = 3)では認めなかった。
C9orf72変異ではTDP-43の異常を伴うがSOD1変異では伴わない事を考えると、stathmin-2の機能喪失はTDP-43による運動神経変性に関与している可能性は確かそうだ。
また、前頭側頭認知症を伴うALS(ALS-FTD)の患者(n = 4)でも同様の所見が確認された。
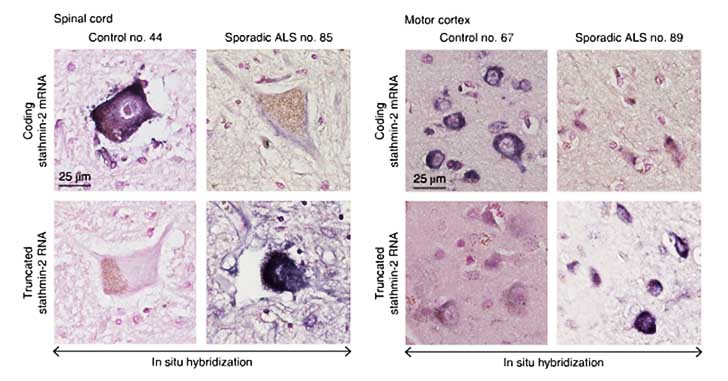
最後に、孤発性ALS患者の運動野および脊髄の組織をin situ hybridizationで調べたところ、ALS患者では、健常人で多く発現している全長のstathmin-2 mRNAが減少し、切断されたstathmin-2のmRNAが増えている像が確認された。
TDP-43の機能喪失による神経軸索再生の障害は、stathmin-2の導入により緩和される
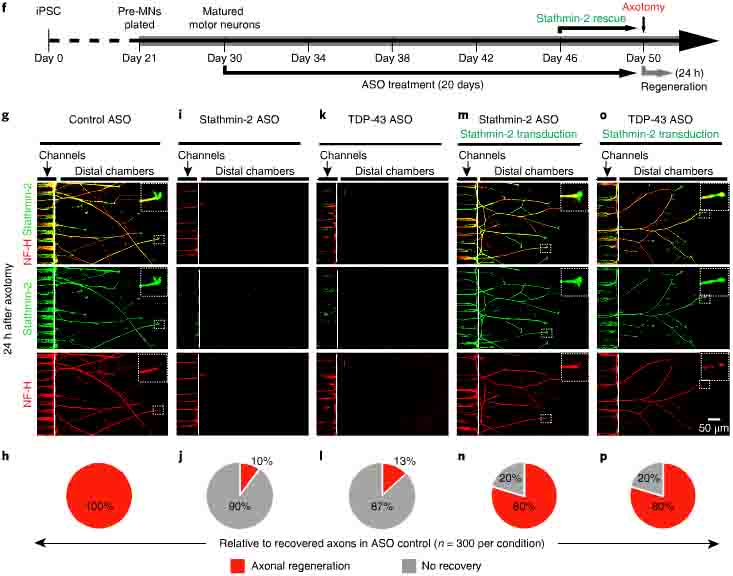
TDP-43とstathmin-2の機能的評価を行うため、著者らは、iPS細胞を運動神経細胞に分化させ、アンチセンスオリゴ(Antisense oligonucleotieds: ASOs)を用いてTDP-43 mRNAとstathmin-2 mRNAの発現を抑制させた。
双方の発現抑制は、運動神経の生存率には影響しなかった。
次に、運動神経前駆細胞をmicrofluidicのデバイスに播種した。軸索を機械的に切断すると、軸索再生がおこるが、この過程でstathmin-2は斑状となり、軸索再生に関与していた。
この時、stathmin-2とTDP-43の発現をASOで抑制すると、神経軸索の再生は、それぞれ10%、13%程度にまで抑制された。
次に、この状態の運動神経にレンチウイルスを用いてstathim-2遺伝子ど形質導入したところ、神経軸索はコントロールと同じくらいまで再生した。
cf. Melamed Ze'ev et.al., Nat Neurosci. 2019 Feb;22(2):180-190. doi: 10.1038/s41593-018-0293-z.
My View
先月のNature Neuroscienceに、同じような内容の論文が抱き合わせで2報掲載されました。(もう一つは、Harvard 大学から、Kilm JR et.al., Nat Neurosci. 2019 Feb;22(2):167-179. doi: 10.1038/s41593-018-0300-4.)
こちらの報告の方のDon Clevelandのラボは、アンチセンスオリゴを得意としていたと思います。ALS以外でも、「ほとんどの神経変性疾患はアンチセンスオリゴで内因性のタンパクを減らせば治療できるよ!」と、以前のシンポジウムでも、ユーモアたっぷりの楽しい話を展開させていました(もちろん、データも十分期待できる内容でした)。
TDP-43はFTLD-UとALS患者の細胞内封入体の構成タンパクとして、2006年に報告(Neumann M et.al., Science, 2006)されました。これまでにTDP-43の様々な機能が解明されてきていますが、ALS/FTLDの病態機序への関与としては、まだまだ分からないことが多いと思います。
一般的には、TDP-43の異常がALS/FTLDの病態を引き起こす機序としては、Loss of functionとGain of toxic functionの両方があると考えられています。
今回はLoss of functionの機序の一部を説明できたことで、大変意義深いと思いますが、機能評価がちょっと弱いかなーと思ったことと、Gain of toxic functionには触れられていないことが気になりました。
先日のScienceにGain of toxic functionの方に迫った研究が掲載されていたので、機会があればそちらの方も紹介したいと思います。