ゴーシェ病の原因遺伝子 GBA1。
GBA1 はリソソーム脂質加水分解酵素グルコセレブロシダーゼ(Glucocerebrosidase, GCase)をコードする遺伝子で [1] 、GBA1のホモ変異でGCaseの活性が低下し、基質のグルコセレブロシドが肝脾、骨髄、脳内などに蓄積し重篤な症状をきたします。
この変異をヘテロで持つキャリアの人達は、ゴーシェ病への罹患は免れますが、晩年パーキンソン病(Parkinson's disease, PD)やレヴィ小体型認知症(Dementia with Lewy bodies, DLB)などのレヴィ小体病(Lewy body disease, LBD)に罹患しやすいことが以前から知られていました [2, 3] 。
そして2011年にGCaseとα-シヌクレイン(α-synuclein, α-syn)の相互作用が報告され [4]、
Parkinson's disease (PD), an adult neurodegenerative disorder, has been clinically linked to the lysosomal storage disorder Gaucher disease (GD), but …

さらにGBA1変異がない孤発性PDでもGCase活性が下がっている事が報告されました [5]。
現在は、Ambroxol など、GCase活性を上げる薬剤のPD治療への臨床試験が進められています。[6, 7, 8, 9]。
この2011年の論文では、GBA1変異でGCaseの基質のグルコセレブロシドが溜まり、それを足場にしてα-Synの凝集が促進される、という事が1つの機序として示唆されていますが、
変異GCaseはそれ以外にも悪さをしているのかもしれません。
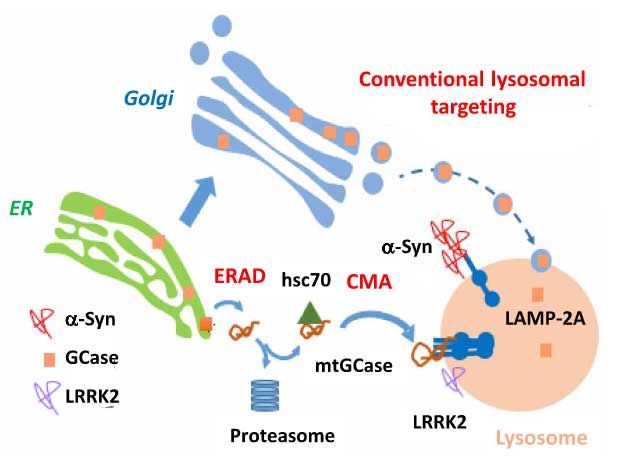
今回、アメリカ・コロンビア大学の Dr. Sulzer, アルベルト・アインシュタイン医科大学の Dr. Cuervo らの研究グループは、「変異GCaseは半分ほどリソソームの表面に異所局在し、これによってシャペロン介在性オートファジー(CMA)によるα-Synの分解が阻害される」という事を報告しました [10]。

変異Gcaseでシャペロン介在性オートファジーを介したα-シヌクレインの分解が阻害される
変異GCaseでは、局在がリソソーム外に変化する
彼らは、SH-SY5Y細胞を使って、
- 野生型(wild-type, WT)
- N370S変異(NS)
- L444P変異(LP)
- D409H(DH)
のGCaseが発現する系を作りました。
また、マウスの初代培養腹側中脳ドパミンニューロンに、
- 野生型(wild-type, WT)
- NS変異
を形質導入しました。
既報通り、mycでタグ付けしたWTのGCaseはリソソーム内(LAMP1+)に入りましたが、変異GCase達ではその割合が減少しており、多くのGCaseが小胞体(endoplasmic reticulum, ER)(BiP)やサイトゾルの中に存在していました。
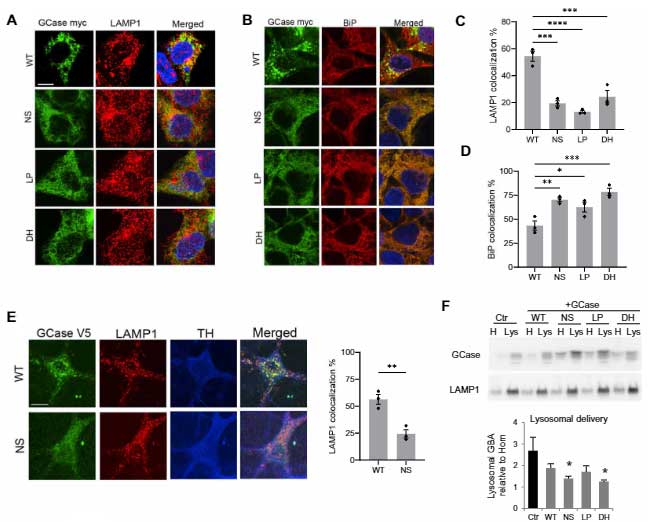
ERやリソソームにある、リソソーム内腔タンパク・カテプシンDや、リソソーム内のリソソーム膜タンパク・LAMP1の発現量には変化がありませんでした。
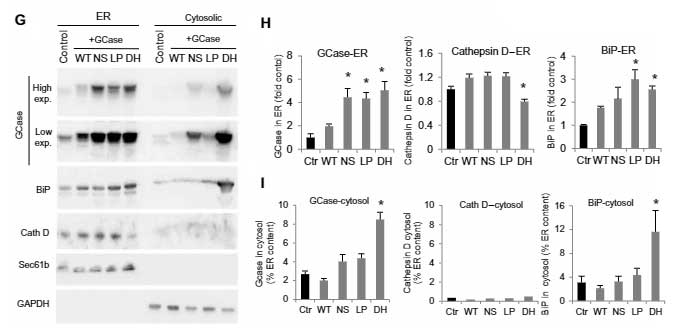
同様の結果は、iPS細胞や
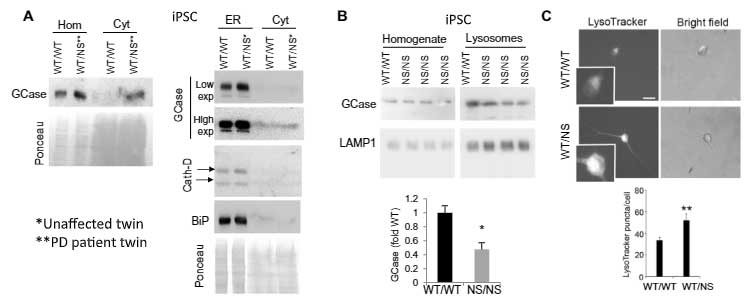
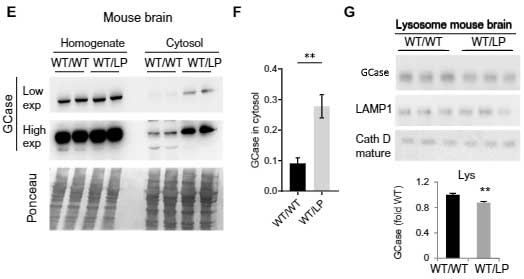
マウス脳等でも観察されました。
異常GCaseはUPSだけでなくCMAを介しても分解される
既報では、変異GCaseはプロテアソームで分解されると言われていますが、著者らは、プロテアソーム以外にリソソームでも分解されるのではないかと考え、WTとGCaseWT/NSのPD患者の線維芽細胞を調べました。
変異GCaseは、プロテアソーム阻害剤のMG-132処置でレベルが上がり、プロテアソームで分解される事を示唆していましたが、
同じくらいの割合でリソソーム阻害剤の塩化アンモニウムやロイペプチン(N/L)でもレベルが上がっており、またER 変異GCaseはリソソームでも分解される事がわかりました。
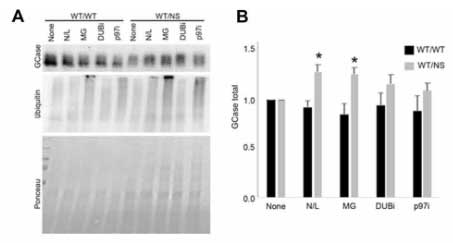
(WTの線維芽細胞では、MG132でもN/Lでも変化がなく、タンパク分解をほとんど受けていない事がわかります。)
先の結果ではGCaseがリソソーム表面に存在していたので、彼らはGCaseがCMAを介してリソソームに取り込まれている可能性を考え、in vitro CMA Assayで調べました。
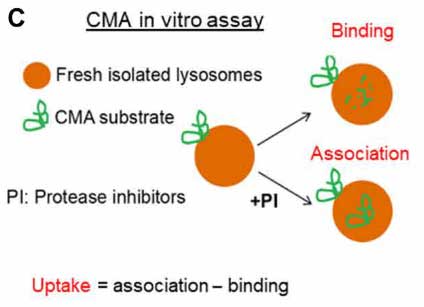
このアッセイでは、リソソームを単離して外からGCaseを処置してどれくらいの量がリソソームに結合/取り込みされるのかをみますが、
通常の「ER→ゴルジ→リソソーム」という経路を通ってリソソーム内に取り込まれている内因性GCaseは、グリコシル化を受けて分子量が高くなっているので、内因性と外因性のGCaseはバンドの高さで見分ける事ができます。
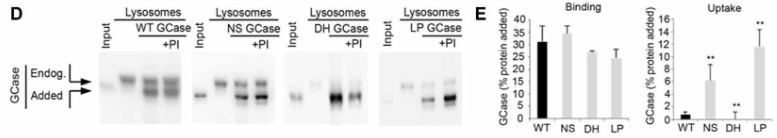
結果、外から加えたGCaseはリソソームの表面に結合するものの、中に取り込まれる割合は、
- NS変異とLP変異は既報の基質と比べて20-30%程
- WTとDH変異は既報の基質と比べて0.5-1.4%程
と、かなり減少していました。
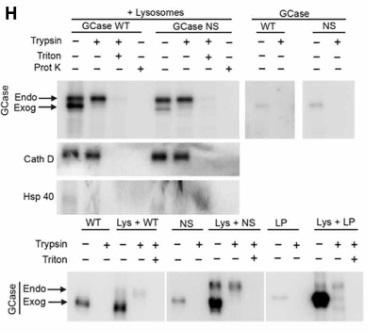
GCaseがリソソームの内側と外側どちらにいるのか調べるため、著者らは
- トリプシン処理
- トリプシン処理+Triton X-100
- プロテイナーゼK
の3種類で処置した所、
内因性のGCaseはトリプシン処置だけでは分解されず、Triton X-100で膜を壊すと分解され、またTritonなしでもリソソームの膜内に侵入できるプロテイナーゼKで処置すると内因性/外因性どちらのGCaseも分解される一方で、
外因性のGCaseは普通にトリプシンのみで分解されたので、
外から加えたGCaseはリソソームの膜の外側に結合していたことが証明されました。
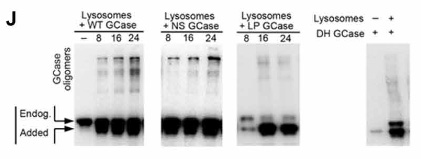
また、フォールドしていない変異GCaseがCMAに結合して留まるのと呼応するように、GCaseのオリゴマー化も促進されていきました。
変異GCaseはCMAを阻害する
著者らは、リソソームの外に結合したGCaseはCMA機能を阻害するのではないかと考えました。
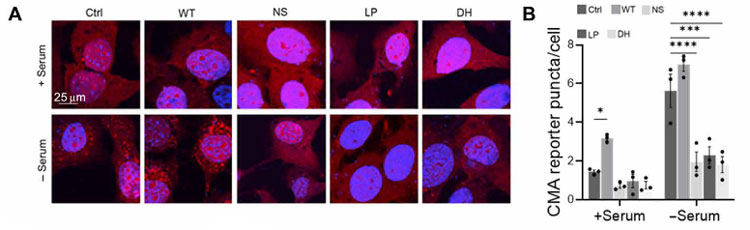
彼らはCMAレポーター(KFFERQ-PA-mCherry1)を、WT, NS, LP, DH GCaseの線維芽細胞に発現させ、血清(-)で負荷をかけた状態でCMAがどうなるか調べました。
結果、コントロールとWT GCaseでは血清(-)でCMAがアップレギュレートされましたが、変異GCaseではその割合が低くなっていました。
CMAターゲットのモチーフ
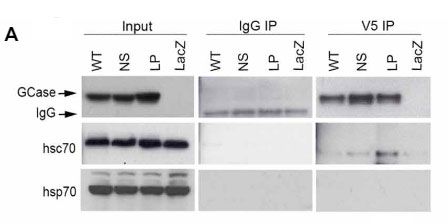
CMAがGCaseをターゲットにするためには、hsc70がモチーフを認識しなければならないわけですが、
彼らは、hsc70で免疫沈降(Immunoprecipitation, IP)して、変異CGaseはhsc70が結合し、WT GCaseには結合していないことを確認しました。
この原因として、きちんとフォールドされたWT GCaseは、モチーフ部分が隠れてhsc70のアクセスを免れている可能性が考えられました。
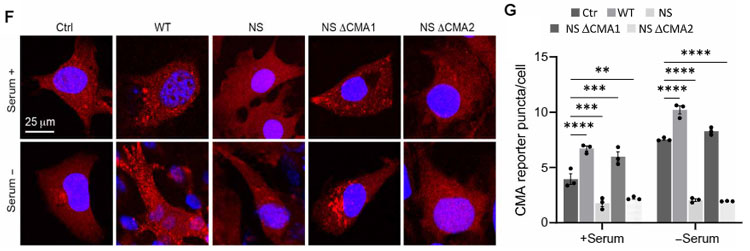
そこで、hsc70が結合する可能性のある2つのモチーフ「256QRDFI260」と「350QSVRL354」を其々欠失させたコンストラクトを入れて検証した所(それぞれ、⊿CMA1と⊿CMA2と表記)、
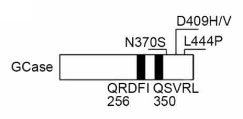
NS変異の⊿CMA1で血清(-)によるCMAアップレギュレーションが確認され、256QRDFI260がhsc70の結合部位であることがわかりました。
変異GCaseはα-Synのリソソームでの分解を妨げる
変異GCaseはリソソームの機能障害をお越し、α-Synの蓄積を引き起こす事が知られていますが [4]、彼らはその原因の1つに今回の現象が関係しているのではないかと考えました。
マウス皮質の初代培養神経細胞(+ドキシサイクリン)で検証した所、α-SynはNS変異GBaseでターンオーバーが滞り、
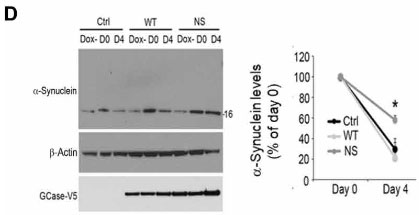
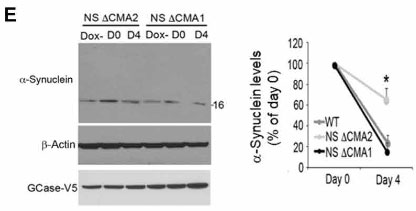
これはNS ⊿CMA1で回復しました。
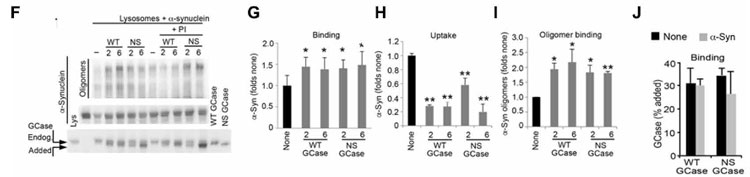
単離したリソソームに外からWTと変異GCaseを加えると、α-Synのリソソーム内への取り込みが阻害され、さらにα-Synのオリゴマー化が促進されました。
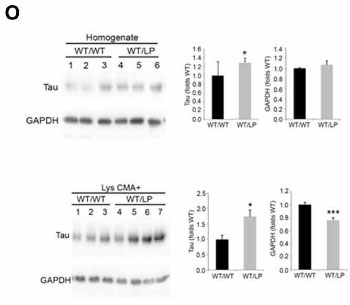
変異GCaseはタウのCMA→リソソーム分解も妨げる
タウはCMAの基質として知られているので、著者らはタウについても同様の検証を行いました。
結果、タウはNS GCaseでCMAを介したリソソームへの取り込みが阻害されており、
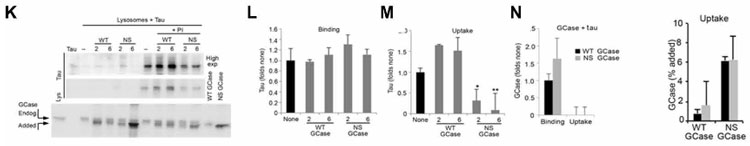
GBA+/L444Pマウス脳内ではタウの蓄積も確認されました。
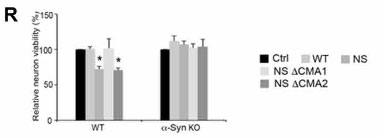
変異GCaseによるCMA阻害はα-Synによる神経障害を引き起こす
WTとα-Synノックアウト(KO)マウスのドパミンニューロンで、WT, NS, NS ⊿CMA1, NS ⊿CMA2を形質導入すると、
NS GCaseで神経生存率が下がり、これは⊿CMA1で回復していました。
このことから、NS変異GCaseによって、α-Synによる神経毒性が生じる可能性が示唆されました。
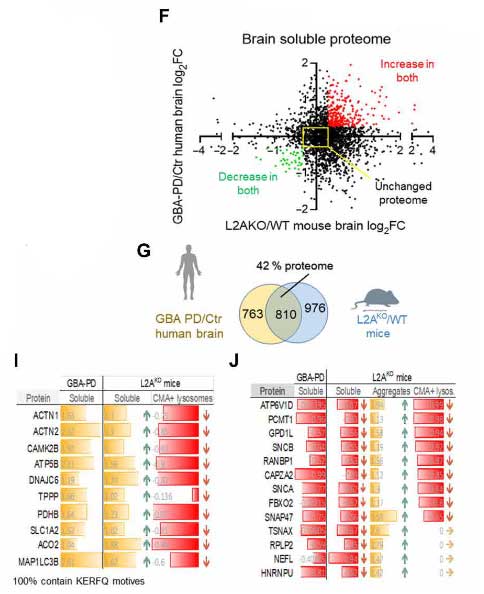
GBA-PD患者さん脳ではCMAの機能障害が起こっている
最後に著者らは、
- 特発性PD(id.PD)
- GBA変異を持つPD(GBA-PD)
- コントロール(Ctr)
の剖検脳でsingle-nucleus RNAシークエンス(snRNA-seq)を行い、CMAコンポーネントの発現レベルがどうなっているか調べました。
ここでは、彼らが以前に確立していたCMA score [11] を用いて評価しましたが、id.PDとGBA-PDではそのCMAスコアが落ちており、CMA機能が落ちている可能性が示唆されました。
また、
- GCaseWT/NS
- GCaseWT/WT
のiPS細胞から分化させたドパミン神経でも、同様の結果が得られました。
GBA-PDで発現が上がっているタンパク達はCMAターゲットモチーフを有しており、CMAが障害されている可能性が示唆されました。

また彼らは、LAMP2AをノックアウトしてCMAが働かなくなるモデルマウス(L2AKO)でもプロテオーム解析を行い、GBA-PDのデータと比較しました。
結果、マウスとヒトと両方で上がっているタンパクは、CMAの基質として知られる糖質や脂質代謝関連タンパクなどが含まれていました。
また、GBA-PDで上がっているタンパクはL2AKOマウスのリソソーム内で上がっており、凝集体を形成していました。
以上の結果から、
- 通常のGCaseはER→ゴルジ→リソソームへ運ばれるのに対して、
- 変異GCaseの一部はERAD機構で細胞質に逆輸送され、 そこでhsc70が結合してCMA機構によって分解されようとするが、
- リソソーム内へはあまり取り込まれず、リソソームの外側に結合したままの状態になる。
- それによりCMAの機能障害が起こって、CMAの基質タンパクの分解が阻害され、
- その中の1つであるα-Synの分解も阻害されて凝集化し、ドパミン神経に対して毒性を持つようになる。
というメカニズムが考えられました。
The most common genetic risk factors for Parkinson’s disease (PD) are a set of heterozygous mutant (MT) alleles of the GBA1 gene that encodes β-glucocerebrosidase (GCase), an enzyme normally trafficked through the ER/Golgi apparatus to the lysosomal lumen. We found that half of the GCase in lysosomes from postmortem human GBA-PD brains was present on the lysosomal surface and that this mislocalization depends on a pentapeptide motif in GCase used to target cytosolic protein for degradation by chaperone-mediated autophagy (CMA). MT GCase at the lysosomal surface inhibits CMA, causing accumulation of CMA substrates including α-synuclein. Single-cell transcriptional analysis and proteomics of brains from GBA-PD patients confirmed reduced CMA activity and proteome changes comparable to those in CMA-deficient mouse brain. Loss of the MT GCase CMA motif rescued primary substantia nigra dopaminergic neurons from MT GCase–induced neuronal death. We conclude that MT GBA1 alleles block CMA function and produce α-synuclein accumulation.

My View
今回何気なく読み始めたのですが……ちょっとハマってしまいました。
原因は、私がCMAとGCaseについてよく理解していなかったからです。
ハマった箇所はFig.3で、ここでは単離したリソソームにWTといくつかの変異GCaseを処置してCMAを介して取り込まれるかどうかをみているのですが、
変異GCaseよりもWT GCaseの方がリソソームの外側に結合しているにもかかわらずリソソーム内にはほとんど取り込まれず、「???」となりました。
その前の2つのFiguresでは「WT GCaseはリソソーム内にいるけど変異GCaseはERや細胞質にいる」という内容だったので、
「WTの方がリソソームに取り込まれて、変異GCaseはリソソーム膜の外側にいるって事じゃないのかな?」と混乱しました。
で、CMAについてざっくり調べて、論文を少し読み進めた結果、私の中では、
「外からふりかけたWT GCaseはむしろ変異GCaseよりもリソソームの外側にくっつく傾向があるけど、ポイントはそこではなくて、
本来のWT GCaseは合成後正常に折りたたまれてゴルジを通って糖鎖付加されてリソソーム内に入ってるから、内因性のWT GCaseは内因性の変異GCaseよりもリソソーム内でちゃんと多くなっているし、
外からふりかけたGCaseはWTだろうと変異だろうとちゃんと折りたたまれてないし、そもそも通常の経路でリソソームへの取り込みを見る系ではないし、
このunfoldedなGCaseをCMAが処理しようとするかどうかがポイントなのか。」
という事で納得しました。
(もし間違ってたら指摘してください。)
あと、GCaseについても、
「本来はリソソームで分解酵素として働くはずだけど、異常GCaseの場合はただのloss of functionだけじゃなくて、それ自体がgain of toxic functionで分解機構を阻害したりする」
という所がちゃんと頭の中で整理されてなくて、時折混乱しました。
2011年の論文 [4] では、
「変異GCaseは合成後すぐにUPSで分解されちゃうので、loss of functionでその基質のグルコセレブロシダーぜがリソソーム内で溜まって、それを足場にα-Synが凝集する」
というストーリーだったと思うんですが、
今回の論文だと、
「それだけじゃなくて、変異GCaseの一部はCMAで分解されるために別経路でリソソームに運ばれて、でもそこで立ち往生しながらCMAの機能自体を阻害してしまう」
という別の機能が提唱された、という事のようです。
で、疑問に思ったのは、
「通常のGCaseも変異GCaseも別経路で同じリソソームに到達するけど、通常版は他のタンパクを分解するためにやってきて、変異版は逆に自分が分解されるためにやってくるんだよね。
じゃあ、通常のGCaseって、なんで他の酵素で分解されないのかな?」
という事。
うまい具合に折りたたまれていて、色々な酵素の襲撃を免れる形になっているのでしょうか?
でも、タウとかα-Synとか、ガチガチに固まった凝集体でも分解しちゃうようなリソソーム内で、全ての切断部分を隠しながら存在するって可能なんでしょうか?
GCaseに限らず、リソソーム内の酵素達って、なんで自分達は分解されずにちゃんと働けるんでしょうか?
いやはや、分解系は個人的に大変興味があるのですが、知識と理解が追いつかなくて凹む事が多いです。
……勉強せねば。
Glossary
タンパク分解機構 [12]
タンパク分解機構には、大きく分けて
- ユビキチン・プロテアソーム系(ubiquitin proteasome system, UPS)
- シャペロン介在性オートファジー(chaperone‐mediated autophagy, CMA)
- マクロオートファジー(macroautopathy)
がある。
イメージとしては、
misfolded / unfolded タンパクはほとんどUPSで分解され、
KFERQモチーフがあるものはCMAで分解され、
凝集体のような大きくて強固な異常タンパクはマクロオートファジーで分解される、
という感じ。
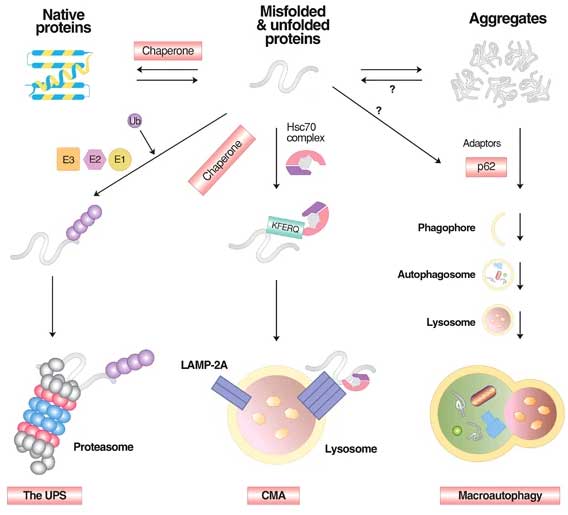
シャペロン介在性オートファジー(chaperone mediated autopathy)[13, 14]
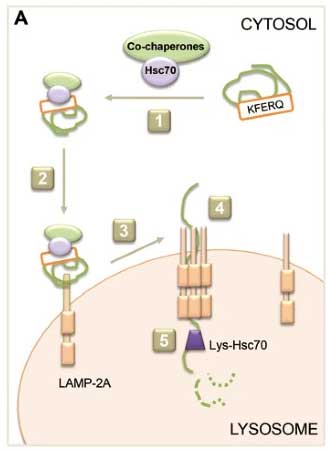
CMAは、複数のステップで成り立っている [13]。
- Hsc70とCo-chaperonesが基質のKFERQモチーフを認識し、
- 基質をリソソームまで運び、
- 基質をリソソームのLAMP-2A受容体に結合させ、
- LAMP-2A受容体が複合体を形成し、
- 基質の折りたたみ構造が解かれ、
- リソソーム膜内側のシャペロン(Lys-Hsc70)アシストされながら基質がリソソーム内に入り、
- 基質がリソソーム内で分解される。
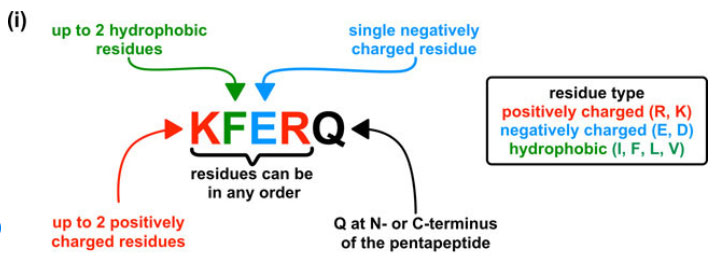
CMAのhsc70が認識するモチーフは、「KFERQ」配列だが、その決まりとして以下のような法則がある [14] 。
- 5つのペプチドで構成され、
- N末かC末にglutamine (Q)があり、
- それ以外は
- 2個までの疎水性アミノ酸(isoleucine(I), phenylalanine (F), leucine (L), valine (V))
- 2個までの陽性荷電アミノ酸(arginine (R), lysine (K))
- 1個の陰性荷電アミノ酸(glutamate (E), asparate (D))
- となっている。
in vitro CMAアッセイ [15]
基質がリソソームに結合/分解される量を定量する方法。
- 細胞からリソソームを単離する。
- 基質と一緒にインキュベーションする。
- リソソームを回収し、SDS-PAGEで、基質の抗体でブロットする。
- リソソームに結合するタンパクは、通常の方法で測定。
- リソソームに取り込まれるタンパクは、プロテイナーゼ阻害剤なしの結果から、プロテイナーゼ阻害剤ありの結果を指し引いて算出する。
※ 今回、GBaseタンパクはdenaturizedして、凝集体はショートスピンで取り除いている。
小胞体分解(endoplasmic reticulum-associated degradation, ERAD)と逆行輸送(retrotranslocation)[16, 17]
小胞体では多くの膜タンパクや分泌タンパクが整合性されているが、正しくフォールドしたタンパクのみが分泌経路にのり、ミスフォールドしたタンパクは小胞体に留められる。
ミスフォールドしたタンパクは、小胞体内に留められて、分子シャペロンやフォールディング酵素の助けを借りて再フォールディングが試みられるが、最終的にフォールディング不能と判断されると、細胞内分解を受けることになる。
この際、小胞体ストレス(ER stress)が惹起され、適応反応である小胞体ストレス反応(unfolded protein response, UPR)の1つとして、異常タンパクが小胞体から細胞質に逆輸送(retrotranslocation)された後に、細胞質に存在するプロテアソームによって分解されるが、この機構をERADと呼ぶ。
References
- Siebert M, Sidransky E, Westbroek W. Glucocerebrosidase is shaking up the synucleinopathies. Brain. 2014 May;137(Pt 5):1304-22. doi: 10.1093/brain/awu002. Epub 2014 Feb 14. PMID: 24531622; PMCID: PMC3999712.
- Do J, McKinney C, Sharma P, Sidransky E. Glucocerebrosidase and its relevance to Parkinson disease. Mol Neurodegener. 2019 Aug 29;14(1):36. doi: 10.1186/s13024-019-0336-2. PMID: 31464647; PMCID: PMC6716912.
- Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, Chen CM, Clark LN, Condroyer C, De Marco EV, Dürr A, Eblan MJ, Fahn S, Farrer MJ, Fung HC, Gan-Or Z, Gasser T, Gershoni-Baruch R, Giladi N, Griffith A, Gurevich T, Januario C, Kropp P, Lang AE, Lee-Chen GJ, Lesage S, Marder K, Mata IF, Mirelman A, Mitsui J, Mizuta I, Nicoletti G, Oliveira C, Ottman R, Orr-Urtreger A, Pereira LV, Quattrone A, Rogaeva E, Rolfs A, Rosenbaum H, Rozenberg R, Samii A, Samaddar T, Schulte C, Sharma M, Singleton A, Spitz M, Tan EK, Tayebi N, Toda T, Troiano AR, Tsuji S, Wittstock M, Wolfsberg TG, Wu YR, Zabetian CP, Zhao Y, Ziegler SG. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med. 2009 Oct 22;361(17):1651-61. doi: 10.1056/NEJMoa0901281. PMID: 19846850; PMCID: PMC2856322.
- Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011 Jul 8;146(1):37-52. doi: 10.1016/j.cell.2011.06.001. Epub 2011 Jun 23. PMID: 21700325; PMCID:PMC3132082.
- Gegg ME, Burke D, Heales SJ, Cooper JM, Hardy J, Wood NW, Schapira AH. Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann Neurol. 2012 Sep;72(3):455-63. doi: 10.1002/ana.23614. PMID: 23034917; PMCID: PMC3638323.
- ClinicalTrials.gov, Ambroxol in Disease Modification in Parkinson Disease (AiM-PD)
- Mullin S, Smith L, Lee K, D'Souza G, Woodgate P, Elflein J, Hällqvist J, Toffoli M, Streeter A, Hosking J, Heywood WE, Khengar R, Campbell P, Hehir J, Cable S, Mills K, Zetterberg H, Limousin P, Libri V, Foltynie T, Schapira AHV. Ambroxol for the Treatment of Patients With Parkinson Disease With and Without Glucocerebrosidase Gene Mutations: A Nonrandomized, Noncontrolled Trial. JAMA Neurol. 2020 Apr 1;77(4):427-434. doi: 10.1001/jamaneurol.2019.4611. PMID: 31930374; PMCID: PMC6990847.
- ClinicalTrials.gov, Ambroxol as a Treatment for Parkinson's Disease Dementia
- Silveira CRA, MacKinley J, Coleman K, Li Z, Finger E, Bartha R, Morrow SA, Wells J, Borrie M, Tirona RG, Rupar CA, Zou G, Hegele RA, Mahuran D, MacDonald P, Jenkins ME, Jog M, Pasternak SH. Ambroxol as a novel disease-modifying treatment for Parkinson's disease dementia: protocol for a single-centre, randomized, double-blind, placebo-controlled trial. BMC Neurol. 2019 Feb 9;19(1):20. doi: 10.1186/s12883-019-1252-3. PMID: 30738426; PMCID: PMC6368728.
- Kuo SH, Tasset I, Cheng MM, Diaz A, Pan MK, Lieberman OJ, Hutten SJ, Alcalay RN, Kim S, Ximénez-Embún P, Fan L, Kim D, Ko HS, Yacoubian T, Kanter E, Liu L, Tang G, Muñoz J, Sardi SP, Li A, Gan L, Cuervo AM, Sulzer D. Mutant glucocerebrosidase impairs α-synuclein degradation by blockade of chaperone-mediated autophagy. Sci Adv. 2022 Feb 11;8(6):eabm6393. doi: 10.1126/sciadv.abm6393. Epub 2022 Feb 9. PMID: 35138901.
- Kirchner P, Bourdenx M, Madrigal-Matute J, Tiano S, Diaz A, Bartholdy BA, Will B, Cuervo AM. Proteome-wide analysis of chaperone-mediated autophagy targeting motifs. PLoS Biol. 2019 May 31;17(5):e3000301. doi: 10.1371/journal.pbio.3000301 Erratu m in: PLoS Biol. 2022Feb 4;20(2):e3001550. PMID: 31150375; PMCID: PMC6561683.
- Patel B, Cuervo AM. Methods to study chaperone-mediated autophagy. Methods. 2015 Mar;75:133-40. doi: 10.1016/j.ymeth.2015.01.003 Epub 2 015 Jan 14. PMID: 25595300; PMCID: PMC4355229.
- Ciechanover A, Kwon YT. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med. 2015 Mar 13;47(3):e147. doi: 10.1038/emm.2014.117. PMID: 25766616; PMCID: PMC4351408.
- Jackson MP, Hewitt EW. Cellular proteostasis: degradation of misfolded proteins by lysosomes. Essays Biochem. 2016 Oct 15;60(2):173-180. doi: 10.1042/EBC20160005. PMID: 27744333; PMCID: PMC5065703.
- Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 2014 Jan;24(1):92-104. doi: 10.1038/cr.2013.153 Epub 2013 Nov 26. PMID: 24281265; PMCID: PMC3879702.
- Tsai B, Ye Y, Rapoport TA. Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat Rev Mol Cell Biol. 2002 Apr;3(4):246-55. doi: 10.1038/nrm780. PMID: 11994744.
- Carvalho P, Stanley AM, Rapoport TA. Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell. 2010 Nov 12;143(4):579-91. doi: 10.1016/j.cell.2010.10.028. PMID: 21074049; PMCID: PMC3026631.